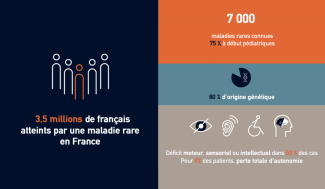
Une maladie est dite rare lorsqu’elle affecte moins d’une personne sur 2 000. Plus de 7 000 maladies rares – dont environ 800 neurologiques – sont décrites, et de nouvelles sont identifiées chaque semaine. Les maladies rares sont généralement sévères, chroniques, handicapantes. La moitié des personnes atteintes présentent des déficits moteurs, sensitifs ou intellectuel et dans 9 % des cas, une perte totale d’autonomie. C’est dans ce contexte que les équipes de recherche de l’Institut du Cerveau et de la Moelle épinière (Institut du Cerveau – ICM) tentent aujourd’hui d’identifier les causes de 14 maladies neurologiques rares afin d’accélérer leur diagnostic et de proposer des traitements adaptés en particulier par thérapie génique.
L’identification de gènes, de réseaux de neurones spécifiques, les études d’imagerie cérébrale, la mise en évidence de marqueurs précoces de la pathologie, sont autant de résultats prometteurs pour le développement de nouvelles approches thérapeutiques. Les pistes d’investigation issues de ces recherches sont d’ores et déjà à l’étude, comme la stimulation cérébrale profonde dans le syndrome de Gilles de la Tourette ou la thérapie génique pour la maladie de Huntington.
Maladie de Huntington, Ataxies spinocérébelleuses : l’espoir et les questionnements des ARN anti-sens
« L’étude de la phase asymptomatique des maladies neurogénétiques rares est un axe majeur de l’équipe. Tous les patients atteints de la maladie de Huntington sont nés avec la mutation génétique. Pourtant les symptômes n’apparaissent que 30 ou 40 ans plus tard. Que se passe-t-il avant ? Finalement, la protéine anormale, elle, s’accumule depuis la naissance dans le cerveau des patients. Quels sont donc les mécanismes de compensation qui permettent de repousser l’apparition des symptômes visibles de plusieurs dizaines d’années ? » explique Alexandra Durr, neurogénéticienne et co-cheffe de l’équipe « Neurogénétique fondamentale et translationnelle ».
Une collaboration active entre la clinicienne-chercheuse et le Dr Sandrine Humbert au GIN à Grenoble qui a montré que des défauts de migration cellulaire étaient déjà présents à l’état fœtal. En revanche, la maladie ne se manifeste pas dès la naissance. L’étude Win-HD qui a étudié la substance blanche du cerveau, très en amont de la maladie, chez des sujets jeunes, ne montre aucune atteinte détectable.
« Il y a des signes d’atteinte cellulaire chez le fœtus, mais aucune atteinte clinique chez l’enfant et l’adolescent et les signes cliniques apparaissent à l’âge adulte. Cela veut dire que ces jeunes ont compensé leur déficit. Comment font-ils cela ? C’est ce que nous cherchons à savoir. Si nous le comprenons, nous pourrons chercher à booster ces processus de compensation pour retarder le développement de la maladie. » poursuit la chercheuse.
Dans la maladie de Huntington comme dans les ataxies spinocérébelleuses dominantes, une protéine anormale s’accumule dans les cellules, déclenchant un phénomène toxique. Pour la première fois depuis 25 ans, la médecine a les moyens de s’attaquer à ces protéines. L’idée est d’envoyer un petit bout de matériel génétique, un ARN anti-sens, qui va bloquer non pas le gène muté en lui-même, mais son ARN messager qui permet à la synthèse de la protéine. L’ARN messager est recyclé en permanence. Le cibler donner la possibilité d’intervenir au moment voulu, de façon ponctuelle, sans modifier définitivement le gène.
Un essai de phase III est en cours avec Roche®, basé sur une injection tous les deux mois d’ARN anti-sens chez 660 patients atteints de la maladie de Huntington. « Une centaine de centres dans le monde se sont réunis pour cet essai. Nous avons inclus tous les patients en à peine 3 mois. C’est exceptionnel ! » précise Alexandra Durr. Si les résultats sont positifs, plusieurs questions resteront encore à élucider : quelle est la meilleure fenêtre pour traiter les patients ? C’est tout l’intérêt des études de la phase asymptomatique de la maladie pendant laquelle le processus pathologique est compensé. De plus, ce traitement cible aussi bien la protéine mutée que la protéine normale, toujours produite chez les patients. Quel effet cela aura-t-il à long terme ?
Dans les ataxies spinocérébelleuses, en particulier dans SCA7, un traitement anti-sens est également en préparation. Une des raisons de l’aggravation de la maladie chez les patients est l’instabilité de la mutation responsable de l’ataxie. Alors qu’elle est extrêmement stable chez le fœtus, la forme de la mutation change complètement d’une cellule à l’autre du cerveau à l’âge adulte. Une grande étude génétique a permis d’identifier des potentiels modificateurs de l’âge de début, partagés entre la maladie de Huntington et des ataxies cérébelleuses, toutes dues à une expansion d’un motif répété dans le gène responsable. Les modificateurs font partie du système de réparation de l’ADN. L’anti-sens que les médecins cherchent à utiliser en ciblant un de ce gènes modificateurs a pour l’objectif de stabiliser l’expansion et donc de limiter l’évolution de la maladie.
Le syndrome du déficit en GLUT-1, un modèle pour la compréhension du métabolisme cérébral
La maladie de De Vivo ou syndrome du déficit en transporteur cérébral de glucose de type 1 (GLUT-1) est une maladie rare touchant environ 800 personnes en France, dont un peu plus d’une centaine serait diagnostiquée. Il existe plusieurs types de transporteurs à la surface des cellules, selon les organes, permettant de faire pénétrer le glucose à l’intérieur de cellules pour produire l’énergie nécessaire à leur fonctionnement. GLUT-1 est particulièrement exprimé dans les globules rouges, les vaisseaux sanguins et les cellules gliales du cerveau, comme les astrocytes qui sont les cellules nourricières du cerveau. Ce transporteur est donc un régulateur fondamental du métabolisme du sucre dans le cerveau.
La maladie de De Vivo est une maladie génétique dite dominante, c’est-à-dire que la mutation d’une des deux copies du gène codant pour le récepteur GLUT-1 suffit à perturber la fonction de ce transporteur. L’anomalie génétique peut être héritée d’un parent, mais le plus souvent il s’agit d’une mutation de novo. L’utilisation altérée du glucose qui résulte de l’atteinte du transporteur GLUT-1 entraine des symptômes très diverses comme des déficiences intellectuelles et des troubles de l’apprentissage chez les enfants, de l’épilepsie, et du fait des changements dans le métabolisme cérébral avec l’âge, l’épilepsie tend à diminuer avec l’âge alors que la composante des mouvements anormaux se majore quant à elle. Les mouvements anormaux peuvent être permanents ou paroxystiques, c’est-à-dire survenir par accès en particulier à la faveur d’un exercice ou d’un jeûne.
Une collaboration entre le Dr Fanny Mochel et la Start-up Metafora a permis de développer un test diagnostique par prise de sang qui détecte la quantité du transporteur au niveau des globules rouges, plus simple et moins invasif que la méthode de diagnostic la plus courante aujourd’hui qu’est la ponction lombaire. Les chercheurs évaluent actuellement, en collaboration avec la Haute Autorité de Santé, le rendu diagnostique de ce test en pratique clinique courante dans une quarantaine de centres hospitaliers français.
Sur le plan thérapeutique, la stratégie thérapeutique de référence consiste à utiliser une autre source d’énergie que le glucose : les corps cétoniques. L’apport de grandes quantités de lipides du régime cétogène permet de les transformer en corps cétoniques, qui sont utilisés par le cerveau de façon très efficace sans passer par le transporteur GLUT-1. Ce régime est très efficace contre les crises d’épilepsie mais semble moins efficace sur les mouvements anormaux. Il est de plus très contraignant pour les patients.
Les chercheurs de l’Institut du Cerveau – ICM ont eu l’idée d’une thérapeutique : une huile synthétique appelée triheptanoïne. Il s’agit d’un acide gras de plus petite taille que les lipides de l’alimentation, il n’a donc pas besoin d’une machinerie complexe pour rentrer et être utilisé dans les cellules et pénètre facilement dans le cerveau. La quantité nécessaire de cette huile est aussi beaucoup plus faible que les corps cétoniques, ce qui la rend beaucoup moins contraignante pour le régime alimentaire des patients. La triheptanoïne a par ailleurs une structure chimique originale qui lui permet d’apporter de l’énergie aux cellules d’une façon plus efficace que les corps cétoniques. Des études récentes conduites à l’Institut du Cerveau – ICM ont montré une diminution de plus de 90% des mouvements anormaux paroxystiques chez les patients déficitaires en GLUT1 traités par triheptanoïne, avec un effet stable dans le temps.
A présent, les axes de recherche sont d’évaluer le bénéfice de la triheptanoïne sur la composante épileptique de la maladie, en particulier chez les enfants, ainsi que d’évaluer la possible transition pour certains patients présentant des symptômes résiduels sous régime cétogène au traitement par triheptanoïne. Plus généralement, ce déficit en GLUT-1 représente un modèle pour comprendre le métabolisme du glucose dans le cerveau, car beaucoup d’autres pathologies neurodégénératives comme la maladie d’Alzheimer ou la maladie de Huntington présente un hypométabolisme cérébral du glucose. Un objectif de Fanny Mochel et de son équipe est d’étudier, grâce à des analyses d’imagerie cérébrale, les effets du traitement sur le cerveau pour mieux comprendre les spécificités du métabolisme cérébral.
Les paraplégies spastiques héréditaires : diagnostic et traitement en ligne de mire
Les paraplégies spastiques sont la deuxième maladie la plus fréquente touchant les neurones moteurs avec une prévalence d’environ 5 patients pour 100 000 personnes. Les neurones moteurs forment un faisceau de fibres allant du cortex moteur à la moelle épinière, responsable de la motricité volontaire et qui dégénère dans les paraplégies spastiques. Ce sont des maladies complexes et très hétérogènes.
Le diagnostic, un enjeu clé pour les paraplégies spastiques héréditaires
Depuis 10 ans, la nouvelle génération de séquençage a révolutionné l’approche de la génétique de ces maladies. Elle a permis d’identifier environ 70 gènes responsables de paraplégies spastiques héréditaires. Malheureusement, malgré tous ces gènes connus, la moitié des cas restent inexpliqués.
La première explication pour expliquer cette difficulté diagnostique vient des méthodes de séquençage génétique. Celles-ci ne séquencent souvent qu’une partie de l’ADN : l’exome. Des informations clés pourraient se trouver dans d’autres parties de l’ADN, les séquences non-codantes, ce qui représente un champ de recherche assez vaste. Ce fut par exemple le cas pour des maladies très proches, les ataxies spinocérébelleuses, dans lesquelles une mutation a été très récemment identifiée dans ces parties non-codantes de l’ADN. Autre explication, les algorithmes qui analysent les séquences ADN s’améliorent de jour en jour. Les chercheurs ont observé qu’en ré-analysant des séquences qui n’avaient pas montré de mutations il y a quelques années avec les algorithmes d’aujourd’hui, ils pouvaient révéler, dans un petit pourcentage de cas, des mutations qui étaient masquées auparavant. Giovanni Stevanin, co-responsable de l’équipe « Neurogénétique fondamentale et translationnelle» est l’un des partenaire d’un projet européen, SOLVE-RD (pour « résoudre les maladies rares »), qui a pour but d’analyser les séquences de 19 000 patients qui avaient été trouvés négatifs lors du premier séquençage de leur ADN, pour tenter de trouver de nouvelles mutations génétiques. Enfin, il reste sûrement des gènes à identifier, et de nouveaux gènes sont régulièrement identifiés par l’équipe de neurogénétique de l’Institut du Cerveau – ICM, qui possèdent un vrai savoir-faire dans ce domaine.
L’équipe de Giovanni Stevanin et Alexandra Durr est à l’origine d’un kit diagnostic par nouvelle génération de séquençage, qui permet de séquencer les 70 gènes en cause dans les paraplégies spastiques héréditaires en un temps record. Développé à l’Institut du Cerveau – ICM avec la plateforme de génotypage-séquençage, ce kit est à présent utilisé en pratique clinique courante à l’unité fonctionnelle de génétique clinique de la Pitié-Salpêtrière, site de référence hospitalier pour les paraplégies spastiques.
Le diagnostic des paraplégies spastiques est un enjeu majeur à l’heure actuelle, car l’objectif sous-jacent est de construire des cohortes de patients suffisantes pour chaque forme de paraplégie spastique afin d’identifier des biomarqueurs, qui permettront de suivre l’effet des futurs essais thérapeutiques.
SPG11 : un exemple d’un aller-retour du patient au laboratoire réussi
Suite à l’identification à l’Institut du Cerveau – ICM d’un gène à l’origine de la forme la plus fréquente des paraplégies spastiques autosomiques récessives – les deux copies du gène doivent être mutées pour que la maladie s’exprime cliniquement-, SPG11, chez des patients, l’équipe « Neurogénétique fondamentale et translationnelle » a développé un modèle expérimental très précis, récapitulant tous les aspects de la pathologie humaine, pour mieux comprendre les mécanismes de la maladie. Giovanni Stevanin, Khalid El-Hachimi et Frédéric Darios ont identifié des accumulations de certains lipides dans les cellules. Cette accumulation de lipides intervient très tôt dans la pathologie, avant l’apparition des symptômes cliniques. Cette découverte a fait naître une idée de thérapeutique dans l’esprit des chercheurs : empêcher l’accumulation de ces lipides dans le cerveau grâce à une molécule thérapeutique déjà utilisée dans une autre pathologie. Les essais conduits dans un modèle expérimental donnent des résultats prometteurs mais le passage à l’homme nécessite d’adapter le traitement avec une autre approche en cours d’étude.
« Notre travail sur SPG11 est un bel exemple d’un aller-retour entre le patient et le laboratoire. Nous avons identifié la mutation chez les patients et établi des corrélations entre gènes et symptômes. Nous avons ensuite développé un modèle expérimental qui nous a permis de détailler le rôle de la protéine codée par le gène SPG11 et l’accumulation de lipides dans les neurones, que nous avons également validé chez les patients. Ceci nous a enfin permis de pointer une cible thérapeutique, que nous essayons à présent d’approfondir pour traiter les patients dans l’avenir. » explique Giovanni Stevanin.
Dystonie et mouvements anormaux rares : une équipe pionnière
L’équipe de Marie Vidailhet et Stéphane Lehéricy à l’Institut du Cerveau – ICM travaille depuis des années sur les maladies du mouvement, et en particulier les dystonies et les mouvements anormaux rares.
Ils ont démontré dans la dystonie une implication forte du cervelet, qui a à la fois une capacité de compensation mais aussi d’altération du mouvement. La dystonie n’est pas due à des lésions dans le cerveau mais à un dysfonctionnement de certains circuits cérébraux. Les chercheurs développent donc des recherches pour trouver des solutions comme la rééducation, la stimulation magnétique transcrânienne, de nouvelles techniques audacieuses de stimulation non-invasives et la stimulation cérébrale profonde pour tenter de rétablir un fonctionnement normal du cerveau et améliorer les symptômes.
« La prise en charge des mouvements anormaux rares et des dystonies se fait à l’hôpital par une consultation dédiée dans notre Centre National de Référence Maladies Rares. Il y a d’abord le diagnostic et l’explication du diagnostic. Mettre en lien le patient avec les associations de patients est très important. Il existe une panoplie de traitements, médicamenteux, chirurgicaux et d’essais que nous pouvons proposer. » explique Marie Vidailhet.
L’équipe de Marie Vidailhet et Stéphane Lehéricy est une équipe pionnière dans le tremblement orthostatique primaire, pour comprendre les anomalies qui sous-tendent ce trouble. Le tremblement orthostatique primaire se caractérise par un tremblement à haute fréquence mais imperceptible par les patients. Il pourrait être lié à une dysfonction du cervelet ou de la moelle épinière. La stimulation non-invasive de la moelle épinière est une piste qu’explore actuellement les chercheurs.
Deux essais récents ont donné des résultats prometteurs dans les mouvements anormaux rares. Dans une dystonie causée par une mutation du gène ADCY5, qui se distingue par spasmes pendant la nuit, Aurélie Méneret et Emmanuel Roze, ont montré que le café, très fort, peu améliorer les symptômes de ces sujets. Dans les dystonies paroxystiques, qui sont également liées à une anomalie génétique, PRRT2, les chercheurs ont étudié les mécanismes, par explorations électrophysiologiques et par imagerie. Là encore, un traitement a pu être développé, basé sur le repositionnement de médicaments antiépileptiques.
« Pour ces maladies, nos espoirs sont énormes. Ces personnes ont un cerveau normal mais sa fonction est altérée avec une dérégulation de l’activité des circuits cérébraux. Nos espoirs sont donc de trouver des méthodes qui régularisent, rétablissent la normalité la fonction du cerveau. Faire que la personne se répare elle-même en l’aidant grâce à des techniques thérapeutiques est l’ambition de notre recherche et, pour certains sujets, déjà applicable dans le quotidien. » conclut Marie Vidailhet
Syndrome Gilles de la Tourette
Le syndrome Gilles de la Tourette est une pathologie neuropsychiatrique et neuro-développementale très hétérogène qui se caractérisent par des tics, des mouvements brusques et répétés involontaires. Ces derniers apparaissent durant l’enfance, autour de 6-7 ans, et sont presque toujours associés par la suite à des symptômes psychiatriques. Une autre particularité de ce syndrome est qu’il se résorbe ou s’améliore spontanément à l’âge adulte chez environ 25% des patients. Pour les 75% restants, les symptômes peuvent se maintenir ou même s’aggraver. Un enjeu dans cette pathologie est donc de comprendre pourquoi certains patients s’améliorent spontanément afin de pouvoir développer des stratégies thérapeutiques pour aider les autres.
Retrouvez la vidéo avec Andreas HARTMANN, coordonnateur du centre de référence Syndrome Gilles de la Tourette :
Les impulsions motrices ne prédisent pas les tics des patients
Cyril Atkinson-Clément (Institut du Cerveau – ICM) et Yulia Worbe (Sorbonne Université/APHP) dans l’équipe « MOV’IT : mouvement, investigations, thérapeutique. Mouvement normal et anormal : physiopathologie et thérapeutique expérimentale» à l’Institut du Cerveau – ICM, montre que le contrôle de l’impulsivité motrice, ce trait qui caractérise la capacité à inhiber un mouvement ou une action déjà commencé, n’est pas corrélé avec les tics chez les patients atteints du syndrome Gilles de la Tourette. Ces résultats, publiés dans la revue Cortex, apportent un nouvel éclairage sur cette pathologie complexe.
Retrouvez l’article en intégralité sur ce lien
La paralysie supranucléaire progressive
Retrouvez l’interview du Pr Jean-Christophe Corvol et du Dr Isabelle Le Ber sur cette page.
Les chercheurs de l’Institut du Cerveau – ICM travaillent sur de nombreuses autres maladies rares : Sclérose latérale amyotrophique, épilepsies génétiques, tumeurs cérébrales, trisomie 21, leucodystrophies… dont plusieurs feront l’objet de communication à l’occasion de prochaines journées dédiées. Continuez de suivre l’Institut du Cerveau – ICM sur ses réseaux sociaux pour être tenu informé des dernières avancées dans ces pathologies.