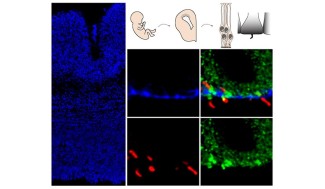
La maladie de Huntington, anciennement appelée chorée de Huntington, est une affection neurologique, rare et héréditaire décrite pour la 1ere fois par Georges Huntington, médecin généraliste en 1872. Elle se manifeste à l’âge adulte par des troubles moteurs, cognitifs et psychiatriques qui s’aggravent progressivement.
-

18 mille personnes sont touchées en France
Il y a environ 10 cas de maladie de Huntington pour 100 000 individus. Elle se déclare entre 35 et 50 ans avec la même prévalence chez les hommes et les femmes. Dans 10% des cas, la maladie se déclare avant 20 ans.
En France, 18 000 personnes sont touchées dont seulement 6 000 présentent actuellement des symptômes et environ 12 000 porteuses du gène muté de la huntingtine restent à ce jour asymptomatiques.
Les causes et les mécanismes biologiques de la maladie de Huntington
La maladie de Huntington est une maladie génétique et héréditaire, due à une expansion d’un motif CAG dans le gène codant pour la protéine huntingtine. Elle se transmet sur un mode autosomique dominant : une copie hétérozygote de l’expansion est suffisante pour développer la maladie plus tard dans la vie. Les descendants des personnes avec cette pathologie ont 50% de risque d’avoir hérité l’expansion.
Les symptômes et le diagnostic de la maladie de Huntington
Les symptômes de la maladie de Huntington se manifestent souvent entre 30 et 50 ans avec l’apparition de troubles moteurs, dont la chorée, des troubles anxieux et dépressifs, surtout remarqués par l’entourage et qui sont accessibles à un traitement symptomatique efficace à l’installation des symptômes.
Les symptômes moteurs sont en général des mouvements anormaux, brusques, saccadés, involontaires des 4 membres, un trouble de l’équilibre et une dysarthrie. D’un point de vue cognitif, on observe chez les patients surtout un ralentissement du traitement des informations, des difficultés de planifier les actions et avec le temps apparaissent des troubles de la mémoire et de concentration.
Ces troubles sont associés très rapidement de troubles comportementaux comme une irritabilité, une anxiété ou une apathie, qui aboutissent à un syndrome sous cortico-frontal.
Le diagnostic initial est basé sur signes présentés par les patients et peut être confirmé par l’analyse génétique dont la famille doit être préalablement informée. La mise en évidence de l’expansion permet aux familles concernées d’avoir recours au conseil génétique pour savoir s’ils sont porteurs ou non de la mutation et par conséquent à un diagnostic prénatal ou préimplantatoire en cas de projet parental.
Les traitements de la maladie de Huntington
Il n’existe pas de traitement curatif de la maladie de Huntington, différentes traitements symptomatiques peuvent être proposées. Des neuroleptiques, inhibiteurs dopaminergiques et des anti-dépresseurs adaptés diminuent les signes moteurs, psychiatriques et les troubles du sommeil. La kinésithérapie et l’orthophonie sont nécessaires et efficaces. Un suivi psychologique est important à la fois pour la prise en charge des symptômes de la maladie et pour l’accompagnement du patient et de ses proches. L’accompagnement social est crucial.
L’équipe "Développement et dysfonctionnement cérébral dans les maladies neurogénétiques", dirigée par Alexandra DURR et Sandrine HUMBERT s’intéresse à des maladies neurogénétiques, les dégénérescences spinocérébelleuses- DSC (les paraplégies...
En savoir plusSoutenez l'Institut du Cerveau
Ce contenu vous a plu et vous a permis de répondre aux questions que vous vous posiez ? N'hésitez pas à soutenir l'Institut du Cerveau.