Les mécanismes physiopathologiques de la SLA à l’origine de la dégénérescence des motoneurones sont encore mal compris. La découverte des mutations génétiques associées à la maladie a permis d’en identifier un certain nombre.
Les mécanismes biologiques de la maladie de Charcot ?
Découverte en 2011, la mutation du gène C9ORF72 est présente dans environ 40% des formes familiales de SLA, 30% des formes familiales de DFT, et 70% des patients atteints des deux pathologies DFT/SLA. En 2006, des agrégations toxiques de la protéine TDP-43 avaient également été identifiés dans ces maladies. On sait aujourd’hui qu’ils sont présents dans 95% des cas de SLA, toutes formes confondues (familiales et sporadiques), et 60% des DFT. La découverte de ces agrégats communs aux deux pathologies a permis de donner une base moléculaire à ce continuum clinique observé depuis longtemps par les médecins. Par ailleurs, tous les porteurs du gène C9ORF72 muté présentent également des agrégats de TDP-43, sans qu’un lien physiologique entre les deux, de même que leurs interactions, ne soient à l’heure actuelle clairement identifiés.

D’après Elisa TEYSSOU et al, Acta Neuropathologica 2013, 125 : 512-522.
Images générées (ou produites ?) par le Pr Danielle SEILHEAN, anatomopathologiste dans l’équipe de Séverine BOILLEE
Une perturbation de l’autophagie ?
Une des raisons suspectées de la présence de ces agrégats toxiques est un défaut du recyclage des déchets à l’intérieur des cellules. Le gène de l’Ubiquiline 2 (UBQLN2) fait partie des gènes impliqués dans le développement de la SLA. Celui-ci est nécessaire à la dégradation des déchets dans les neurones. Une étude conduite par plusieurs équipes de l’Institut du Cerveau en collaboration avec l’Université de Limoges s’est penchée sur les mutations de UBQLN2. Grâce à des études fonctionnelles, les chercheurs ont mis en évidence que l’une des voies de dégradation cellulaire, l’autophagie, était altérée chez les patients porteurs d’une mutation de l’Ubiquiline 2.
Plus d’informations : https://institutducerveau-icm.org/fr/actualite/sla-de-nouvelles-decouvertes/
Une inflammation chronique
Il est connu depuis longtemps que les agrégats protéiques dans le cerveau, comme les plaques amyloïdes dans la maladie d’Alzheimer ou l’a-synucléine dans la maladie de Parkinson, sont sources d’inflammation, mais cela reste à élucider dans le cas de TDP-43.
Le rôle de la microglie, les cellules immunitaires résidentes du système nerveux, dans la maladie est aujourd’hui bien admis. Les motoneurones spinaux affectés dans la SLA ont la particularité d’être entourés à la fois par des cellules microgliales dans la moelle épinière et par des macrophages périphériques dans le nerf qui contiennent la partie du motoneurone sortant de la colonne vertébrale pour connecter le muscle à la périphérie. Or, le rôle des macrophages était jusqu’à récemment sujet à débat. En 2020, l’équipe de Séverine Boillée à l’Institut du Cerveau a montré pour la première fois un rôle important des macrophages périphériques dans la progression de la SLA. Comme les microglies, ces derniers génèrent un état inflammatoire chronique délétère pour les motoneurones. L’intérêt des macrophages est qu’ils seraient plus faciles à cibler, depuis la périphérie, que les cellules microgliales, dans le système nerveux central.
Plus d’informations : https://institutducerveau-icm.org/fr/actualite/sclerose-laterale-amyotrophique-sla-maladie-de-charcot/
À l’Institut du Cerveau
Modéliser les interactions cellulaires de la SLA grâce aux cellules des patients
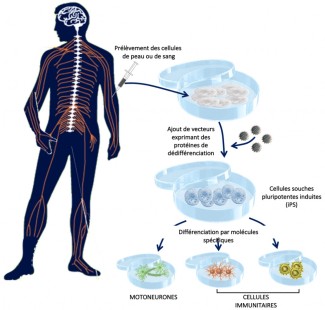
L’objectif de Delphine BOHL dans l’équipe de Séverine BOILLEE est de modéliser la maladie grâce aux cellules souches pluripotentes induites, humaines (iPSC). Cette technologie de pointe permet de générer tout type de cellules, incluant des motoneurones ou des cellules immunitaires à partir de cellules de peau (biopsie) de patients. Les iPSC ont deux grandes capacités qui sont celles d’être capables de se multiplier à l’infini et de se différencier en n’importe quel type cellulaire de l’organisme pour peu qu’elles soient exposées aux bons signaux. Ces nouveaux modèles cellulaires sont un outil précieux qui permet pour la première fois d’avoir accès à des neurones humains de patients. La première étape est de pouvoir caractériser très précisément les motoneurones obtenus à partir des cellules iPSC de patients, d’abord dans des cas génétiques, où la mutation à l’origine de la SLA est connue, puis dans des cas sporadiques afin d’éventuellement identifier des mécanismes communs. La deuxième étape est de réunir dans un même milieu, très contrôlé, les motoneurones et les cellules immunitaires pour modéliser leurs interactions. À termes, ces modèles permettraient également de tester l’efficacité des molécules thérapeutiques.
Protocole d’obtention de cellules souches pluripotentes induites (iPS) et leurs dérivés