Une étude conduite par l’équipe du Pr. Bertrand Fontaine (APHP, Sorbonne Université) à l’Institut du Cerveau – ICM, en collaboration avec le centre de référence des pathologies neuromusculaires dirigé par le Pr. Bruno Eymard à la Pitié-Salpêtrière a mis en évidence de nouvelles mutations dans un gène, GFPT1, responsable d’une forme particulière de syndromes myasthéniques congénitaux (SMC). Au-delà d’une meilleure compréhension des mécanismes sous-jacents à ces pathologies, ils suggèrent une possible évolution de la classification de ce syndrome particulier et une meilleure prise en charge des patients.
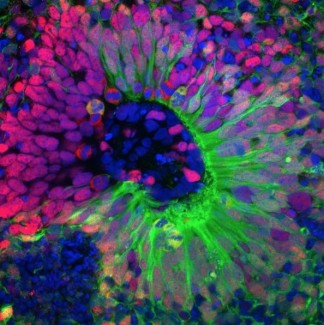
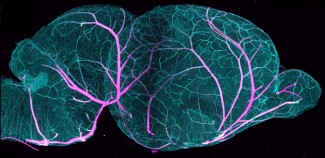
Les syndromes myasthéniques congénitaux sont un ensemble de pathologies rares, cliniquement et génétiquement hétérogènes, caractérisées par un dysfonctionnement de la jonction neuromusculaire (JNM), zone de communication entre le nerf moteur et le muscle. Les patients atteints par ces pathologies présentent des symptômes moteurs, liés à des anomalies de la transmission nerveuse entre les nerfs et les muscles, et caractérisés majoritairement par une fatigabilité musculaire dont l’intensité varie au cours du temps.
Les mutations à l’origine des SMC sont variées touchant des gènes codant des protéines essentielles pour la formation, la maturation et le maintien de la JNM. Actuellement, 31 gènes à l’origine des SMC ont été identifiés, dont le gène ubiquitaire GFPT1. Ce gène code pour une enzyme nécessaire à la glycosylation des protéines, mécanisme indispensable au bon fonctionnement de la JNM. Dans la littérature, 52 mutations dans ce gène ont été répertoriées comme responsable de SMC. Dans les syndromes myasthéniques congénitaux liés à ces mutations, la distribution de la faiblesse musculaire se situe au niveau des muscles proximaux, c’est à dire les muscles des épaules et des bras pour les membres supérieurs ainsi que les muscles des hanches et des cuisses pour les membres inférieurs.
Dans 9 familles de patients SMC, issus de la cohorte française suivis depuis plus de 20 ans par le centre de référence des pathologies neuromusculaires, l’équipe du Pr. Fontaine a identifié 12 mutations dans le gène GFPT1 parmi lesquels 9 n’avait jamais été décrites. Une étude clinique rétrospective de ces patients a montré une évolution vers des symptômes de type myopathique, c’est-à-dire avec une faiblesse musculaire accentuée, s’accompagnant d’une inefficacité des traitements les plus couramment prescrits chez ses patients.
Une analyse de la jonction neuromusculaire chez 3 patients possédant des mutations dans le gène GFPT1, a validé, pour la première fois, que l’atteinte de la jonction neuromusculaire n’est pas limité à un seul compartiment de celle-ci mais à deux (pré et post-synaptique). De plus, une analyse moléculaire du muscle de 4 patients Français a mis en évidence que des voies de signalisation autres que celle directement liée à la mutation de GFPT1, peuvent être perturbées. Ces voies sont notamment impliquées dans certaines formes de myopathie (GNE-myopathie), pouvant expliquer la progression de la maladie chez ces patients.
Dans leur ensemble, ces résultats permettent une compréhension plus globale des mécanismes mis en jeu dans cette forme particulière de SMC « ubiquitaire » qui pourrait être considérée comme une myopathie avec une atteinte de la synapse plutôt qu’un syndrome myasthénique congénital, apportant ainsi un regard nouveau pour le diagnostic clinique, l’identification et la prise en charge des patients.
Sources
https://pubmed.ncbi.nlm.nih.gov/28712002/
Bauché S, Vellieux G, Sternberg D, Fontenille MJ, De Bruyckere E, Davoine CS, Brochier G, Messéant J, Wolf L, Fardeau M, Lacène E, Romero N, Koenig J, Fournier E, Hantaï D, Streichenberger N, Manel V, Lacour A, Nadaj-Pakleza A, Sukno S, Bouhour F, Laforêt P, Fontaine B, Strochlic L, Eymard B, Chevessier F, Stojkovic T, Nicole S.
J Neurol. 2017 Aug; 264(8):1791-1803.