Les paraplégies spastiques héréditaires représentent un groupe hétérogène de pathologies et partagent des similitudes cliniques avec d’autres maladies neurodégénératives, dont la sclérose latérale amyotrophique (SLA).
Pour la première fois, Khalid H. El Hachimi et ses collaborateurs, dans l’équipe d’Alexis Brice, ont montré des similitudes neuropathologiques (au niveau des lésions du système nerveux) entre la paraplégie spastique de type 11 et la SLA. Cette découverte va permettre aux cliniciens d’effectuer un diagnostic précis et précoce devant une SLA « atypique », via une recherche de mutation du gène SPG11. D’autre part, la compréhension des mécanismes impliqués dans ces pathologies ouvrent la voie au développement de nouvelles thérapeutiques ciblées.
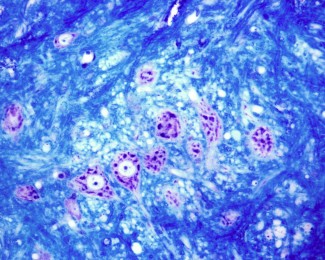
Les paraplégies spastiques héréditaires représentent un groupe hétérogène de maladies sur les plans clinique, génétique et neuropathologique. La forme héréditaire autosomique récessive la plus fréquente est causée par une mutation du gène SPG11. Les signes cliniques s’installent progressivement et sont caractérisés par des troubles de la marche très invalidants dus à une raideur, ou spasticité, des membres inférieurs. Ce tableau clinique peut être compliqué par différents symptômes qui sont partagés avec d’autres maladies neurologiques comme la sclérose latérale amyotrophique, les neuropathies ou les ataxies cérébelleuses. Sur le plan physiopathologique, cette maladie est caractérisée par une dégénérescence du faisceau cortico-spinal (les neurones sont situés dans le cortex moteur et leurs axones se projettent vers la moelle épinière) qui contrôle la motricité volontaire.
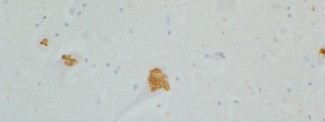
Pour la première fois, Khalid H. El Hachimi et ses collaborateurs, dans l’équipe d’Alexis Brice, ont montré des similitudes neuropathologiques entre la paraplégie spastique de type 11 et la SLA. Grâce à l’étude de cerveaux post-mortem de patients, les chercheurs ont observé des lésions (ubiquitine et P62) au niveau de la moelle épinière et du bulbe rachidien similaires à celles rencontrées dans la SLA. Ces inclusions particulières dans motoneurones sont aussi associées à une accumulation de produits lipidiques. La protéine codée par le gène SPG11 appelée spatacsine est impliquée dans le transport vésiculaire, le transport axonal, la régulation de l’homéostasie calcique et dans le recyclage des lysosomes. La perte de fonction de la spatacsine entrainerait un défaut de recyclage des lysosomes et une accumulation intra neuronale de produits non dégradés qui pourrait être associée à la mort neuronale.
Ces travaux illustrent des similitudes neuropathologiques entre les paraplégies spastiques et la SLA et mettent en exergue le continuum physiopathologique de la dégénérescence des motoneurones. Ces résultats contribuent aussi à une meilleure compréhension des dérégulations lysosomales et pourraient ouvrir de nouvelles pistes pour le développement de solutions thérapeutiques.