Une étude conduite par l’équipe de Stéphanie Baulac met en évidence, pour la première fois chez des patients atteints d’épilepsies focales associées à des malformations cérébrales, le développement d’une mutation du gène DEPDC5 présente uniquement au niveau du foyer épileptogène dans le cerveau, et entrainant la perte totale de fonction du gène. Les résultats sont publiés dans la revue The Journal of Clinical Investigation.
Les épilepsies focales sont localisées dans une zone restreinte du cerveau. Le gène DEPDC5 est un gène majeur des formes familiales de ce type d’épilepsie. Il intervient en amont de la cascade de signalisation mTOR qui contrôle notamment la prolifération et la croissance des cellules et régule le développement cérébral. Dans ces formes d’épilepsie, le gène est muté de façon hétérozygote, c’est à-dire qu’une seule des deux copies du gène porte la mutation.
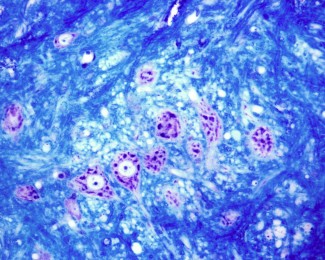
Ces épilepsies peuvent être associées à des malformations du cerveau, les dysplasies corticales focales (FCD). L’épilepsie focale comme les dysplasies sont dites « à foyer variable » c’est-à-dire qu’elles peuvent survenir dans différentes régions du cerveau et ce même au sein d’une même famille. Ces épilepsies sont souvent résistantes aux médicaments antiépileptiques, le retrait par chirurgie du foyer épileptique – la zone du cerveau responsable des crises d’épilepsie – reste alors la seule solution thérapeutique pour les patients.
Pourquoi certains patients épileptiques présentent-ils une malformation corticale et d’autres pas ? Comment expliquer cette diversité de foyers épileptogènes ?
Pour répondre à ces questions, Théo Ribierre et ses collègues ont séquencé un panel de gènes appartenant à la voie mTOR, une cascade de signalisation régulant le développement cérébral impliquée dans les épilepsies focales, dans des tissus cérébraux post-opératoires de patients épileptiques.
Ils ont identifié pour la première fois une seconde mutation dans le gène DEPDC5, qui a pour conséquence l’inactivation de l’unique copie fonctionnelle restante du gène. Il s’agit d’une mutation somatique, présente de façon mosaïque, c’est à dire seulement dans quelques cellules du cerveau, et qui est apparu au cours du développement in utero. Ce type de mutation conduisant à l’absence totale de la protéine dans un nombre restreint et localisé de cellules pourrait expliquer l’apparition des malformations du cortex.
Ils ont également mis en évidence un gradient de mutation au sein de la lésion : le cœur de la lésion qui contient le foyer épileptogène contient jusqu’à 10% des cellules porteuses de cette seconde mutation tandis que dans le tissu dit « péri-lésionnel », soit autour du foyer, ce chiffre tombe à 0,3% des cellules nerveuses.
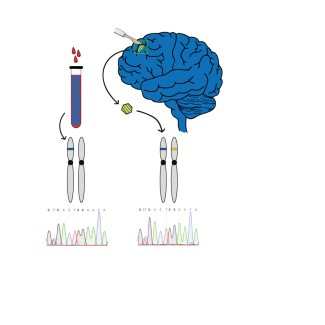
Identification d’une mutation germinale hétérozygote de DEPDC5 présente dans toutes les cellules de l’organisme (ADN extrait de sang et de la malformation corticale), et d’une mutation somatique de DEPDC5 sur le second allèle (conduisant à une inactivation totale du gène) retrouvée uniquement dans 10% des cellules du cœur de la malformation corticale.
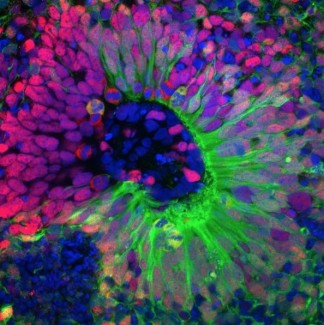
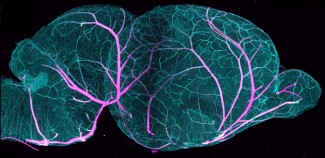
Pour mieux comprendre les mécanismes pathologiques qui conduisent à la fois à la malformation mais aussi à la genèse des crises d’épilepsie, il est nécessaire de développer des modèles récapitulant au mieux la pathologie humaine. Dans la présente étude, l’équipe de Stéphanie Baulac a développé un nouveau modèle murin : une inactivation focale et mosaïque de Depdc5 dans le cerveau.
Les chercheurs ont induit des mutations du gène DEPDC5 dans un nombre restreint de cellules progénitrices de neurones corticaux à l’aide de l’outil CRISPR/Cas9, qui permet de modifier spécifiquement des gènes, et de la technique d’électroporation, permettant de rentrer précisément dans certaines cellules. Le gène DEPDC5 n’était donc plus fonctionnel dans les cellules filles issues de ces cellules progénitrices.
Dans ce modèle, ils retrouvent les défauts histologiques qui caractérisent la dysplasie corticale focale chez l’humain. Les analyses de vidéo électroencéphalogramme ont également montré la présence de crises d’épilepsie spontanées et létales. Enfin, Charlotte Deleuze, sur la plateforme d’électrophysiologie (CELIS E-phys), a pu mettre en évidence des variations des caractéristiques électrophysiologiques des neurones dans lesquels DEPDC5 n’était plus fonctionnel, pouvant contribuer à l’épileptogénèse dans ce modèle.
Ce modèle, qui récapitule donc bien la pathologie humaine, apporte de nouvelles informations sur la fonction de DEPDC5 et pourra à terme permettre de tester de nouveaux candidats-médicaments pour contrôler les épilepsies focales associées à des malformations corticales.
Sources
https://pubmed.ncbi.nlm.nih.gov/29708508/
Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, Muraca G, Roussel D, Leguern E, Miles R, Baulac S. J Clin Invest. 2018 Apr 30