La plupart des maladies neurodégénératives, de la maladie d’Alzheimer à la maladie de Parkinson en passant par la sclérose latérale amyotrophique, partagent des caractéristiques communes avec les maladies à prions.
Ces différentes maladies neurodégénératives auraient pour origine l’accumulation dans le cerveau de protéines anormales, mal repliées et toxiques pour les neurones. Ces protéines pathologiques se propagent de proche en proche d’un neurone à l’autre et se répandent ainsi dans le tissu cérébral conduisant, à terme, à la mort des neurones. Ce processus de propagation est appelé phénomène « prion-like » en référence aux mécanismes décrits dans les maladies à prions. Le « modèle prion » est donc un modèle de premier choix pour comprendre les mécanismes biologiques responsables des protéinopathies.
La 4ème journée d’actualité sur les maladies à prions et autres protéinopathies se tiendra le jeudi 10 décembre 2015 à l’Institut du Cerveau – ICM. Elle est organisée par le Centre National de Référence des Agents Transmissibles Non Conventionnels coordonné par Stéphane Haïk et par la Cellule d’aide à la prise en charge des maladies de Creutzfeldt-Jakob coordonnée par Jean-Philippe Brandel, avec le soutien de l’Institut National de Veille Sanitaire et du Ministère de la Santé.
Cette journée est l’occasion de faire le point sur les maladies à prions et sur les recherches menées à l’Institut du Cerveau – ICM par l’équipe de Stéphane Haïk et Marie-Claude Potier.
Une maladie neurodégénérative
Les maladies à prions, dont la maladie de Creutzfeld Jakob est la forme la plus fréquente, sont des maladies rares. Elles se caractérisent par une démence à laquelle s’ajoutent des signes neurologiques (troubles visuels, troubles de la coordination…). Une fois les premiers symptômes apparus, l’évolution est rapide et conduit au décès en quelques mois. A ce jour, aucun traitement de référence ne peut être proposé.
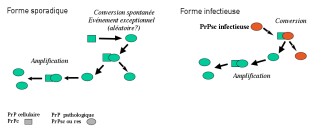
Dans la grande majorité des cas (85 %), la maladie apparaît de façon aléatoire, on parle de maladie sporadique. Dans 15 % des cas, elle peut être due à des mutations génétiques et dans de très rares cas, elle peut être transmise lors de procédures médicales ou d’exposition à des tissus infectés par un agent infectieux (par exemple l’agent de l’encéphalopathie bovine spongiforme qui entraîne la maladie de la vache folle).
Une accumulation de protéines anormales
La protéine prion ou PrP est présente dans la plupart de nos cellules. Bien que très conservée entre les espèces, son rôle est pourtant mal connu. Dans les neurones des personnes malades, les protéines prions adoptent une mauvaise conformation, deviennent résistantes à la dégradation et s’agrègent entre elles : on parle alors de protéines prions « scrapie », PrPsc. Les dépôts de protéines se multiplient et s’accumulent alors à l’intérieur et à l’extérieur des neurones entraînant leur dysfonctionnement, et, à terme, leur dégénérescence. La propagation des prions repose sur leur capacité à interagir avec la forme normale de la protéine prion, d’en changer la conformation et de la convertir en forme pathologique.
Prions, Alzheimer, Parkinson et les autres : même combat
L’équipe de Stéphane Haïk et Marie-Claude Potier à l’Institut du Cerveau – ICM étudie conjointement la maladie d’Alzheimer et les maladies à prions, du fait desanalogies biologiques qu’elles partagent. Elle s’intéresse, entre autres, aux mécanismes moléculaires impliqués dans la propagation des prions, et plus précisément au phénomène de conversion des protéines normales PrP vers des protéines toxiques mal conformées PrPsc qui s’accumulent, se propagent et entrainent la mort des neurones.
Comprendre et mieux diagnostiquer
Les chercheurs ont développé des méthodes d’amplification des protéinesmal conformées in vitro afin d’observer le processus de ≪misfolding » (mauvais repliement des protéines) et de comprendre son rôle dans les mécanismes de mortneuronale. Ces méthodes permettent, d’une part d’étudier les mécanismes depropagation et de modéliser les barrières de transmission de ces agents, et, d’autre part de développer des approches diagnostiques ayant d’excellentessensibilité et spécificité et de tester des moléculesthérapeutiques.
Transférer la technologie
Ce phénomène de propagation ayant été mis en évidence dans d’autres maladies neurodégénératives, des études sont en cours pour transférer cette technologie aux autres protéinopathies (maladies neurodégénératives, caractérisées par l’accumulation anormale de certaines protéines)du système nerveux central, comme la maladie d’Alzheimer (agrégation des peptides β-amyloïdes et de la protéine tau) et la maladie de Parkinson (agrégation de l’α-synucléine). Sa compréhension représente un enjeumajeur pour tenter de le bloquer et de trouver des traitements adaptés à ces pathologies.
Modéliser la propagation des prions au laboratoire
Afin d’étudier la propagation des prions et les conséquences de leur réplication sur la survie et les fonctions des neurones, l’équipe de Stéphane Haïk et Marie-Claude Potier a développé différents modèles spécifiques. Par exemple, le premier système cellulaire permettant de propager différentes souches humaines de prions a été obtenu. Il constitue un outil précieux pour la recherche de molécules thérapeutiques. En effet, l’action des traitements anti-prions varie souvent en fonction des souches, il est donc fondamental d’évaluer leur efficacité vis-à-vis des prions isolés directement chez les patients atteints.
Identifier une nouvelle cible thérapeutique
Les modèles cellulaires mis au point par l’équipe ont également été précieux dans une étude collaborative avec le groupe d’Odile Kellerman et de Benoit Schneider à l’université Paris-Descartes qui a permis de proposer une nouvelle cible thérapeutique commune aux maladies à prions et à la maladie d’Alzheimer. Ce travail a mis en évidence le rôle clef d’une enzyme, PDK1, dans l’accumulation de protéines pathologiques, caractéristique des maladies à prions et de la maladie d’Alzheimer.
Dans les conditions normales, la protéine à l’origine des peptides β-amyloïdes et la protéine prion sont clivées par l’enzyme TACE, présente à la surface des neurones. Les protéines résultant du clivage par TACE ne sont pas toxiques pour le cerveau et jouent même un rôle protecteur pour les neurones.
Dans les neurones infectés par les prions pathogènes comme dans les neurones de patients souffrant de la maladie d’Alzheimer, TACE n’est plus localisée à la surface des cellules, mais est séquestrée à l’intérieur des neurones. L’absence de TACE entraîne un défaut du clivage physiologique de la protéine prion entière non pathologique (PrPc) ou de la protéine précurseur des peptides amyloïdes (APP) ce qui a pour conséquence la production de protéines anormales toxiques.
C’est une autre protéine, appelée PDK1, qui est responsable de la séquestration de TACE à l’intérieur des neurones. En bloquant l’activité de PDK1 dans des modèles expérimentaux, les chercheurs arrivent à bloquer la production de protéines toxiques β-amyloïdes ou prions.
PDK1 représente donc une cible thérapeutique prometteuse pour de futurs traitements à la fois pour la maladie d’Alzheimer et pour les maladies à prion.
La mise en place d’essais cliniques
La rareté de la maladie de Creutzfeldt-Jakob (1-2 nouveaux cas par million d’habitants), la distribution et l’évolution subaigüe de la maladie ont longtemps limité la mise en place d’essais cliniques contrôlés c’est-à-dire dans lesquels l’efficacité d’une nouvelle thérapeutique est comparée à un placebo.
Le plus large essai clinique contrôlé d’envergure internationale (France et Italie) jamais réalisé dans cette maladie a testé l’efficacité de la doxycycline en 20014. Mené en France par des chercheurs de l’Institut du Cerveau – ICM (Jean-Philippe Brandel et Stéphane Haïk), l’étude a ouvert de nouvelles perspectives concernant la mise en place d’essais thérapeutiques contrôlés dans la maladie de Creutzfeld-Jakob.
En France, afin de pouvoir proposer à tout patient suspect et quel que soit son lieu de prise en charge une inclusion dans l’essai avec un délai raisonnable, un dispositif original de recrutement et d’ouverture en urgence de centres investigateurs a été mis en place. En deux ans, ce dispositif a permis de recruter 71 patients dans 49 centres investigateurs dont 27 ont été ouverts en urgence.
Si aucun effet significatif du traitement n’a été mis en évidence, cette étude a apporté des enseignements majeurs pour l’évaluation des futurs traitements qui pourront être proposés dans la maladie de Creutzfeld-Jakob :
Des essais contrôlés versus placebo sont réalisables dans cette maladie
La construction d’essais multinationaux est requise pour permettre le recrutement d’un nombre suffisant de patients dans cette maladie rare et rapidement mortelle.
La procédure originale de recrutement des patients et d’ouverture des centres investigateurs en urgence avec des procédures administratives accélérées mise en place en France pourrait servir d’exemple
En s’appuyant sur cette première expérience internationale et selon l’évolution des données scientifiques, la mise en place d’un futur essai européen sera discutée.
Le saviez-vous ?
Les prions sont à ce jour les seuls agents infectieux dénués de matériel génétique (ADN ou ARN), contrairement aux agents transmissibles conventionnels que sont les virus, les bactéries et les parasites
C’est en 1982, que le neurologue américain, Stanley B. Prusiner, a émis l’hypothèse que l’agent infectieux responsable était en fait une protéine et a créé le terme prion (contraction du terme « proteinaceous infectious particule »). Cette hypothèse, tout à fait originale et « hérétique », est maintenant admise. Ses travaux ont été récompensés par le prix Nobel de médecine en 1997.