A l’occasion de la journée mondiale de la sclérose latérale amyotrophique (SLA) ou maladie de Charcot, l’Institut du Cerveau revient sur une dimension clé de la maladie, particulièrement étudiée par ses équipes : la neuroinflammation. Au-delà de mieux comprendre les mécanismes de la maladie, l’étude de la composante immunitaire pourrait ouvrir de nouvelles pistes thérapeutiques.
Les macrophages périphériques : une piste prometteuse
Les motoneurones spinaux affectés dans la SLA ont la particularité d’être entourés par deux types de cellules immunitaires : des cellules microgliales dans la moelle épinière et par des macrophages périphériques au niveau du nerf, la partie du motoneurone sortant de la colonne vertébrale pour se connecter au muscle en périphérie. Le rôle de ces cellules immunitaires est de défendre l’organisme lors d’une infection mais aussi de contribuer à la cicatrisation des tissus lésés. Les cellules microgliales (ou microglie) sont un sous-groupe de macrophages localisés dans le système nerveux central (moelle épinière et cerveau).
Alors que le rôle de la microglie dans la dégénérescence des motoneurones dans la SLA est aujourd’hui admis, le rôle des macrophages issus de la circulation dans ce mécanisme était encore controversé. L’équipe de Séverine Boillée (Inserm) a mis en évidence la capacité des macrophages périphériques à influencer, de la périphérie, à la fois la réponse des cellules microgliales dans le système nerveux central et la dégénérescence des motoneurones, avec un retard d’apparition des symptômes de la maladie et un allongement significatif de la durée de vie dans le modèle murin.
Les auteurs de ces travaux, publiés dans Nature Neuroscience, concluent pour la première fois, à un rôle important des macrophages périphériques dans l’évolution de la sclérose latérale amyotrophique (SLA), ouvrant ainsi la voie à de nouvelles approches thérapeutiques pour les patients. Ces résultats constituent aujourd’hui une piste de traitement prometteuse puisqu’elle permettrait de cibler les motoneurones directement depuis la périphérie.
Plus d’information : https://institutducerveau-icm.org/fr/actualite/sclerose-laterale-amyotrophique-sla-maladie-de-charcot/
Modéliser les interactions microglies-neurones grâce aux cellules souches
Les cellules souches pluripotentes humaines ou cellules iPS ont deux grandes capacités qui sont celles d’être capables de se multiplier à l’infini et de se différencier en n’importe quel type cellulaire de l’organisme pour peu qu’elles soient exposées aux bons signaux. Ainsi, à partir d’un prélèvement de cellules de peau des patients, les chercheurs développent ces cellules iPS pour ensuite les différencier en différents types de cellules du cerveau, les neurones et les cellules microgliales par exemple. Ces nouveaux modèles cellulaires sont un outil précieux qui permet pour la première fois d’avoir accès à des neurones de patients.
Delphine Bohl (Inserm) cherche à modéliser la maladie grâce à ces cellules. L’objectif est non seulement de pouvoir caractériser très précisément les motoneurones obtenus à partir des cellules iPS de patients, mais également de réunir dans un même milieu, très contrôlé, les motoneurones et les cellules immunitaires pour modéliser leurs interactions. À termes, ces modèles permettraient également de tester l’efficacité des molécules thérapeutiques.
Identifier les causes génétiques de la SLA
10 % des cas de SLA sont des formes dites familiales avec une origine génétique. Stéphanie Millecamps (CNRS) cherche à identifier tous les points de départ génétiques possibles de la maladie dans les cas familiaux et les formes à début précoce. Bien que ces derniers soient multiples, ils pourraient conduire à des voies communes menant à la dégénérescence du motoneurone. Ces études permettent de proposer aux familles qui le souhaitent un diagnostic, et d’identifier les patients éligibles pour les essais thérapeutiques utilisant des oligonucléotides anti-sens dirigés contre la protéine SOD1 ayant d’ores et déjà montré des effets prometteurs.
Un travail collaboratif européen a permis l’identification d’un nouveau gène impliqué dans la SLA. Nommé TBK1, il est impliqué dans l’élimination des déchets à l’intérieur des cellules et dans la régulation de l’inflammation, ce qui démontre l’importance de ces processus cellulaires pour la survie des motoneurones. Les travaux de Christian Lobsiger (Inserm) sur ce gène cherchent à disséquer le rôle spécifique de ce gène dans les motoneurones et les cellules immunitaires dans le contexte de la SLA.
Plus d’information : https://institutducerveau-icm.org/fr/actualite/sla-de-nouvelles-decouvertes/
C9ORF72 : point de départ pour comprendre certains mécanismes dans la SLA
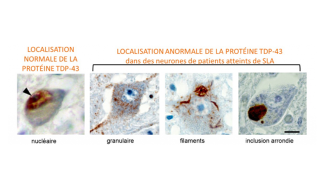
La SLA et les dégénérescences fronto-temporales (DFT) appartiennent à un même spectre de maladies pouvant avoir une cause génétique commune, dont la plus fréquente est une mutation du gène C9ORF72. Découverte en 2011, elle est présente dans environ 40% des formes familiales de SLA, 30% des formes familiales de DFT, et 70% des patients atteints des deux pathologies DFT/SLA. En 2006, des agrégations toxiques de la protéine TDP-43 avaient également été identifiés dans ces maladies. On sait aujourd’hui qu’ils sont présents dans 95% des cas de SLA, toutes formes confondues (familiales et sporadiques), et 60% des DFT. La découverte de ces agrégats communs aux deux pathologies a permis de donner une base moléculaire à ce continuum clinique observé depuis longtemps par les médecins. Par ailleurs, tous les porteurs de la mutation C9ORF72 présentent également des agrégats TDP-43, sans qu’un lien physiologique entre les deux, de même que leurs interactions, ne soient à l’heure actuelle clairement identifiés.
L’objectif du groupe de Morwena Latouche (EPHE) dans l’équipe « Neurogénétique fondamentale et translationnelle » est de comprendre comment l’agrégation de TDP-43 et la mutation C9ORF72 interagissent pour provoquer certains mécanismes de la DFT/SLA. Dans une revue récemment publiée dans Frontiers in Neuroscience, Julie Smeyers, Elena Banchi et Morwena Latouche, ont dressé un bilan des connaissances sur cette mutation et ses implications dans la pathologie.
La mutation du gène C9ORF72 a plusieurs conséquences délétères pour les cellules. Elle conduit à trois types d’effets : une perte de fonction, avec une baisse de la transcription du gène et un double « gain de fonction », par la formation d’un ARN et d’une protéine anormale. Chez les patients, la mutation est hétérozygote, c’est-à-dire que seule une des deux copies du gène est mutée. Ils produisent donc partiellement une protéine C9ORF72 normale mais aussi des ARN et protéines anormaux. En supprimant totalement la protéine dans des modèles expérimentaux, les chercheurs ont mis en évidence une inflammation très importante dans l’ensemble de l’organisme. Par ailleurs, il est connu depuis longtemps que les agrégats protéiques dans le cerveau, comme les plaques amyloïdes dans la maladie d’Alzheimer ou l’a-synucléine dans la maladie de Parkinson, sont sources d’inflammation dans le cerveau, mais cela reste à élucider dans le cas de TDP-43.
L’équipe de l’Institut du Cerveau s’intéresse aujourd’hui au potentiel des agrégats de TDP-43 à générer de l’inflammation et à l’impact de la mutation de C9ORF72, qui sensibilise probablement le système immunitaire, à agir en synergie pour augmenter la réaction inflammatoire délétère au niveau du système nerveux. Leurs résultats pourraient identifier de nouvelles voies à l’origine de la mort des neurones du système nerveux dans les DFT/SLA et permettraient de mieux comprendre le rôle des différents acteurs de ces pathologies.