Des équipes AP-HP viennent de mettre en évidence le premier gène à l’origine d’un défaut de développement de la principale structure cérébrale responsable de la communication entre les deux hémisphères du cerveau. Ces travaux menés par Christel Depienne, chercheur dans l’équipe Institut du Cerveau – ICM « Bases moléculaires, physiopathologie et traitement des maladies neurodégénératives », à l’hôpital de la Pitié-Salpêtrière AP-HP, en collaboration avec le groupe de recherche clinique de Delphine Héron (Hôpital Pitié-Salpêtrière) et les équipes australiennes de Paul Lockhart (Melbourne) et Linda Richards (Brisbane), font l’objet d’une publication dans la revue Nature Genetics, le 27 février 2017.
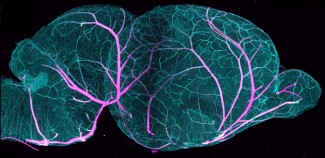
Les deux hémisphères cérébraux sont reliés entre eux et communiquent grâce au corps calleux, une sorte de pont qui permet le passage des informations et participe aux processus de mémoire et apprentissage notamment. Il arrive que certaines personnes naissent sans corps calleux, ce que l’on appelle l’agénésie du corps calleux qui touche un nouveau-né sur 4 000. Il s’agit de l’anomalie cérébrale viable la plus fréquente.
Ce trouble du développement cérébral peut être associé à une déficience intellectuelle de sévérité variable, ou ne se manifester par quasiment aucun symptôme. La détection de cette malformation, est le plus souvent réalisée en période prénatale (échographie au cours du 2nd trimestre de grossesse). Elle constitue alors un véritable challenge pour prédire le devenir cognitif de l’enfant à naître. Dans d’autres cas (patients adultes ou n’ayant pas eu de suivi de grossesse), elle peut être découverte de manière fortuite lors d’un examen IRM réalisé pour une autre cause (bilan de migraine par exemple)
« La découverte du gène DCC nous permet de mieux comprendre les causes d’agénésie du corps calleux et le développement normal et pathologique du cerveau. Elle pourrait avoir un impact majeur dans le cadre du diagnostic prénatal, notamment en permettant de pronostiquer si l’enfant à naître va souffrir ou non d’une déficience intellectuelle ».
Christel Depienne et ses collaborateurs se sont intéressés à une certaine forme d’agénésie calleuse, l’agénésie calleuse isolée, sans déficience intellectuelle associée.
Leur étude, menée chez neuf familles différentes dont quatre avec des personnes atteintes sur plusieurs générations a mis en évidence, pour la première fois, des mutations d’un gène spécifique, le gène DCC, héritées de façon dominante.
La protéine codée par ce gène est un récepteur à la netrin 1 et joue un rôle clé dans le guidage axonal lors du développement cérébral. Elle permet aux axones, prolongements des neurones, de passer d’un côté du cerveau à l’autre et d’assurer ainsi la liaison entre les deux hémisphères.
Le gène DCC avait déjà été associé à une autre pathologie, celle des mouvements en miroir congénitaux, qui affecte la capacité des patients à effectuer un mouvement différent des deux mains.