Les leucodystrophies regroupent une centaine de maladies génétiques rares et se caractérisent par une atteinte de la substance blanche du système nerveux central (cerveau et moelle épinière) avec ou sans altérations de la myéline du système nerveux périphérique.
On estime aujourd’hui entre 150 et 300 le nombre d’enfants par an qui naissent atteints de leucodystrophie en France.
Les causes et les mécanismes biologiques des leucodystrophies
La majorité des leucodystrophies sont génétiques et à ce jour plus de 200 gènes ont été identifiés dans ces pathologies. Le mode de transmission des mutations causales dans la famille ainsi que la fonction du gène muté entraînent des tableaux cliniques différents.
Les leucodystrophies sont causées par des anomalies génétiques appelées mutations dans des gènes qui codent pour des protéines jouant un rôle clé dans la synthèse ou le maintien de la myéline (une substance qui sert à isoler, à protéger les axones des neurones comme une gaine isolante autour de fils électriques et qui joue un rôle important dans la propagation rapide de message nerveux d’un neurone à l’autre).
En effet, la myéline est une membrane ultraspécialisée composée de protéines et des lipides qu’il faut produire et renouveler tout au long de la vie. Des mutations dans un gène impliqué dans une de ces étapes au sein des cellules gliales compromettent les cycles de synthèse et/ou de maintien de la myéline, et par conséquence entrainent une diminution de la quantité de myéline ou une altération des fonctions de la myéline. Toute dégradation de la myéline entraine une souffrance du neurone qui peut alors dégénérer aboutissant à une perte de communication entre le cerveau et la périphérie et l’apparition de symptômes moteurs et sensoriels.
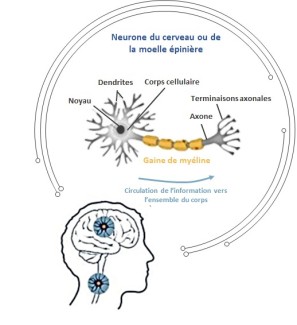
Il existe des leucodystrophies dites « hypomyélinisantes », c’est-à-dire dans lesquelles la production de myéline est insuffisante dès la naissance et des leucodystrophies « démyélinisantes » au cours desquelles on observe une dégradation de la myéline existante. Ces dernières sont en général plus sévères et d’évolution plus rapide.
Les leucodystrophies sont en général des maladies neurodégénératives progressives, et peuvent se manifester dès la petite enfance, l’adolescence ou à l’âge adulte.
Les symptômes et le diagnostic des leucodystrophies
Les leucodystrophies compromettent la fonction de la gaine de myéline. Cela provoque une perte de communication entre les neurones et l’apparition de symptômes moteurs, cognitifs, psychiatriques, et/ou sensoriels.
Les symptômes présentés par les patients diffèrent selon le type de leucodystrophie et degré de sévérité de l’atteinte. Cependant, les signes cliniques communs et les plus fréquents sont, d’un point de vue moteur, une spasticité (raideur des muscles), une ataxie (manque de coordination fine des mouvements volontaires), des troubles de la parole et des mouvements anormaux comme une dystonie (contraction involontaire des muscles) ou des tremblements. Ces troubles moteurs sont souvent associés à des difficultés de contrôle de la vessie.
Les symptômes cognitifs peuvent entrainer selon l’âge des retards du développement psychomoteur, des difficultés scolaires ou professionnelles voire une démence. Les troubles du comportement ou de l’humeur sont également fréquents. Des troubles de la vision et/ou de l’audition sont enfin possibles, en particulier chez les enfants.
Le diagnostic des leucodystrophies est basé en premier lieu sur l’analyse de l’imagerie cérébrale (IRM) qui va permettre de mettre en évidence les anomalies de la myéline et de classer les leucodystrophies en forme hypomyélinisante ou démyélinisante. Le diagnostic plus précis du type de leucodystrophies repose sur des dosages biochimiques pour certaines leucodystrophies et des analyses génétiques dans tous les cas. Lorsque le diagnostic est posé, une enquête familiale (conseil génétique) est proposée afin de déterminer les apparentés à risque de développer ou transmettre la maladie à leur descendance. Il y a souvent des retards de diagnostic des leucodystrophies chez l’adulte car elles sont confondues avec la sclérose en plaque, la maladie acquise de la myéline la plus fréquente et la mieux connue des neurologues. En France, des centres experts nommés Centre de Référence de leucodystrophies rares existent et une de leurs missions est d’aider à un meilleur diagnostic des leucodystrophies ainsi que de proposer une prise en charge adaptée.
Les traitements des leucodystrophies
Pour certaines leucodystrophies, il existe des traitements dédiés, en particulier la greffe de moelle osseuse pour l’adrénoleucodystrophie liée à l’X (X-ALD,) leucodystrophies avec axones sphéroïdes et cellules gliales pigmentées ALSP et certaines formes de leucodystrophie métachromatique. La greffe doit être réalisée à un stade précoce de la maladie pour être efficace. Parfois, des traitements médicamenteux sont possibles, comme dans la xanthomatose cérébrotendineuse. Plusieurs thérapies géniques sont également en développement dans les leucodystrophies.
Par ailleurs, dans toutes les leucodystrophies, des traitements symptomatiques sont possibles, médicamenteux et/ou de rééducation, afin d’améliorer certains symptômes comme la spasticité ou les douleurs.
En 2019, le Pr Fanny MOCHEL, neurologue (hôpital de la Pitié-Salpêtrière, APHP) et chercheuse dans l’équipe MOV’IT des Prs Stéphane LEHERICY et Marie VIDAILHET à l’Institut du Cerveau a réalisé une transplantation de cellules souches hématopoïétiques chez une patiente atteinte d’ALSP à progression rapide. Le suivi de cette patiente sur 30 mois a montré une régression des symptômes et des lésions cérébrales associées à la maladie.
3 types de leucodystrophies étudiées à l’Institut du Cerveau
À l’Institut du Cerveau, les recherches se concentrent sur 3 types de leucodystrophies parmi les plus fréquentes :
• Les adrénoleucodystrophies liées à l’X (ALD)
• Les leucodystrophies métachromatiques (MLD)
• Les leucodystrophies avec axones sphéroïdes et cellules gliales pigmentées (ALSP)
L’équipe de Violetta ZUJOVIC travaille sur les mécanismes de démyélinisation et re-myélinisation dans la sclérose en plaques à partir des cellules du système immunitaire. Comme le montrent les succès de la greffe de moelle, le système immunitaire joue un rôle clé dans certaines leucodystrophies. En collaboration avec le Prof. Fanny MOCHEL et le Dr Caroline SEVIN, Violetta ZUJOVIC étudie désormais le rôle du système immunitaire dans les leucodystrophies afin d’identifier de nouvelles pistes thérapeutiques.
L’équipe du Prof. MOCHEL s’intéresse aux régulations métaboliques des cellules gliales et comment des traitements métaboliques peuvent ralentir voire arrêter certains processus neurodégénératifs. Récemment, son équipe a participé à un essai thérapeutique international qui a montré qu’une molécule ciblant le métabolisme énergétique et l’inflammation dans X-ALD pouvait diminuer le risque de formes cérébrales de la maladie. Des travaux sont en cours pour déterminer si ce traitement peut être une alternative à la greffe de moelle dans X-ALD.
L’équipe du Dr SEVIN utilise la thérapie génique pour corriger l’anomalie génétique en cause à des stades présymptomatiques (avant le début des symptômes) d’enfants atteints d’X-ALD, leucodystrophie métachromatique et maladie de Krabbe.
Les adrénoleucodystrophies liées à l’X (ALD)
Ces maladies touchent 1 nouveau-né sur 17.000 dans le monde. Il s’agit de la leucodystrophie la plus fréquente. L’ALD associe une démyélinisation du système nerveux central et périphérique à un dysfonctionnement des glandes surrénales (insuffisance surrénalienne), des glandes situées au-dessus des reins qui sécrètent certaines hormones telles l’adrénaline.
L’ALD est causée par des mutations du gène ABCD1 situé sur le chromosome X. La protéine ATP-Binding cassette (ABCD1 a pour rôle de dégrader les acides-gras à très longue chaine (AGTLC) à l’intérieur des cellules. Lorsque cette protéine n’est plus fonctionnelle à cause d’une mutation, les AGTLC s’accumulent dans les tissus et deviennent toxiques. Le taux d’AGTLC dans le sang est un marqueur de la maladie qui aide au diagnostic.
Les hommes atteints d’XALD transmettent la maladie à leur filles mais jamais à leurs fils. Les femmes atteintes ont un risque de transmission de la maladie à leurs enfants de 50%.
Il existe 3 tableaux cliniques décrits pour l’ALD qui diffèrent par les symptômes et la population de patients.
Forme | Age de survenue | Symptômes | Traitements |
CALD. ALD cérébrale de l’enfant, de l’adolescent et de l’adulte. Manifestation la plus grave de l’ALD. Elle se caractérise par une dégradation neurologique rapide. La majorité des hommes atteints d’ALD sont à risque de développer une atteinte cérébrale contre moins de 1% des femmes. | C-CALD 3-10 ans Ado-CALD 11-21 ans A-CALD > 18 ans | Troubles attentionnels, visuels, auditifs, psychiatriques associés à des troubles de la marche et de la coordination | Traitements des symptômes. En début de la maladie, une greffe de cellules souches peut être proposée. |
AMN.Adrénomyéloneuropathie. Atteinte chronique et progressive de la myéline centrale et périphérique. Fréquente chez les femmes atteintes de leucodystrophie et chez 100% des hommes atteints. | AMN entre 20 et 40 chez les hommes Entre 40 et 60 ans chez les femmes | Troubles de la marche, de l’équilibre, vésico-sphinctériens. Paralysie légère des membres inférieurs associée à des contractures musculaires. | Traitements des symptômes, spasticité, troubles urinaires et sphinctériens et douleurs neuropathiques. |
ISRN. Insuffisance surrénalienne basse. Cette atteinte se caractérise par une destruction progressive des glandes surrénales. Cette atteinte est retrouvée dans moins de 1% des femmes atteintes de leucodystrophie et dans presque 100% des hommes. | Entre 3 et 10 ans Tous les âges | Le premier signe visible de cette atteinte est un bronzage spontané du patient, la mélanodermie. | Traitement par supplémentation hormonale |
Les leucodystrophies métachromatiques (MLD)
Les leucodystrophies métachromatiques sont des maladies génétiques autosomiques récessives rares qui touchent 1 nouveau-né sur 45 000 et qui peuvent se développer à l’enfance, à l’adolescence ou à l’âge adulte.
Elles sont majoritairement causées par des mutations dans le gène ARSA qui code pour une protéine responsable de la dégradation des lipides appelés sulfatides essentiels pour la formation et le maintien de la myéline centrale et périphérique.
Les formes infantiles de la maladie se développent avant 30 mois et entrainent des symptômes comme une faiblesse musculaire (hypotonie), des troubles du langages, des déficits de motricité. Au fur et à mesure de l’évolution de la maladie apparaissent une spasticité, des crises convulsives, des troubles de la vision et de l’audition.
Les formes juvéniles apparaissent entre 30 mois et 16 ans et entrainent des troubles du comportements et de l’équilibre.
Les formes adultes qui apparaissent après 16 ans induisent des changements de personnalité, une augmentation de l’émotivité associés à des troubles moteurs.
La prise en charge thérapeutique doit-être globale et dans certains cas une greffe de cellules souches peut être envisagée des stades précoces d’évolution.
Les leucodystrophies à sphéroïdes axonaux et cellules gliales pigmentées (ALSP).
Ces leucodystrophies aussi appelées leucoencéphalopathies, sont dues à une mutation dans le gène CSF1-R qui code pour un récepteur présent à la surface des cellules microgliales, cellules immunitaires résidentes du système nerveux central. Ce sont des maladies autosomiques dominantes c’est-à-dire que les enfants d’une personnes atteintes ont un risque de 50% d’être malades. Elles touchent de 10 à 25 % des adultes atteints de leucodystrophies.
L’âge de survenue de ces leucodystrophies est en moyenne 40 ans.
Elle affecte les hommes et les femmes et se présente surtout par une spasticité rapidement progressive et/ou des troubles cognitifs et psychiatriques. Les premiers signes de la maladie surviennent généralement après 30 ans, mais la maladie a une pénétrance incomplète.
Elle se caractérise par une perte progressive de la mémoire, des changements de personnalité, des troubles moteurs associés parfois à une perte de jugement et une désinhibition sociale.
Il n’existe pas à ce jour de traitement spécifique des ALSP. Les risques secondaires liés à cette maladie sont à surveiller comme les infections récurrentes de type pneumonies ou infections urinaires.
La maladie de Pelizaeus Merzbacher
La maladie de Pelizaeus Merzbacher est due à des mutations du gène PLP1 qui code pour une protéine présente dans l’oligodendrocyte qui participe à la synthèse de la myéline. Cette maladie touche surtout les enfants avec des retards de développement important qui impactent tout à la fois les fonctions motrices et cognitives. Les femmes porteuses de mutations du gène PLP1 peuvent également développer des symptômes à l’âge adulte, en particulier une spasticité et des troubles urinaires.
Projets à l’Institut du Cerveau
2 projets collaboratifs associant 2 équipes de recherche sont en cours :
Équipe "Plasticité et régénération de la myéline" - Institut du Cerveau. Porteur de projet Violetta ZUJOVIC, chercheuse INSERM.
Équipe Mov'It - Institut du Cerveau. Porteur de projet Pr Fanny MOCHEL, neurologue APHP.
Le projet Matrix
Le projet MATTRIX concerne l’étude des profils d’activation des macrophages dans l’X-ALD.
Dans cette maladie une inflammation aigue et chronique est observée due à l’activation de cellules immunitaires les macrophages.
Les macrophages sont des cellules sanguines (globules blancs) dont le rôle est d’assurer la défense de l’organisme contre des agents pathogènes comme les virus et les bactéries. Ils participent avec d’autres cellules immunitaires comme les lymphocytes à la réaction inflammatoire.
L’objectif du projet MATTRIX est de comprendre comment les mutations du gène ABCD1 influence les capacités intrinsèques des macrophages à s’activer et à s’orienter vers des profils inflammatoires ou anti-inflammatoires. Les résultats de ce projet pourraient aboutir à une prise en charge individualisée des patients avec une meilleure prédiction de l’évolution de la maladie orientant les traitements de 1ere intention.
Projet Inspire
Le projet INSPIRE porte sur 3 types de leucodystrophies (ALSP, MLD et ALD) et a pour objectifs de :
• Mieux comprendre l’inflammation observée dans ces pathologies afin d’identifier de nouvelles cibles thérapeutiques.
• Contribuer à l’élaboration de critères biologiques fiables pour orienter la prise en charge des patients le plus précocement possible vers une transplantation de cellules souches, seule thérapie efficace sur l’évolution des leucodystrophies actuellement
• Valider un nouveau traitement non invasif pour la leucodystrophie cérébrale (CALD)
• Trouver de nouveaux biomarqueurs pour la détection précoce de la neuroinflammation et mieux comprendre les mécanismes biologiques des leucodystrophies en ciblant la réponse anti-inflammatoire
L’équipe vise à comprendre les mécanismes neuro-inflammatoires et métaboliques impliqués dans la sclérose en plaques et les leucodystrophies, en particulier le rôle du métabolisme cellulaire et des cellules immunitaires dans la progression des...
En savoir plusSoutenez l'Institut du Cerveau
Ce contenu vous a plu et vous a permis de répondre aux questions que vous vous posiez ? N'hésitez pas à soutenir l'Institut du Cerveau.