Une étude menée par l’équipe de Stéphanie Baulac met en évidence des mutations somatiques dans divers types cellulaires chez des patients atteints de dysplasie corticale focale de type 2. Cette maladie cause des épilepsies pharmaco-résistantes dont la principale option thérapeutique est actuellement la chirurgie. A l’aide de séquençage d’ARN de cellule unique sur des tissus postopératoires de patients, les chercheurs ont identifié des mécanismes pathogéniques liés aux mitochondries et à la sénescence cellulaire, spécifique à chaque type cellulaire. Cette découverte ouvre la voie à de nouvelles stratégies thérapeutiques ciblées et offre un espoir pour les patients inéligibles à la chirurgie. Les résultats sont publiés dans la prestigieuse revue Nature Neuroscience.
Vers un traitement de la dysplasie corticale focale de type 2
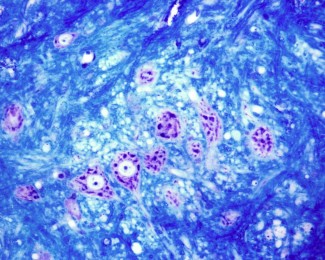
La dysplasie corticale focale de type 2 (DCF2) est une malformation cérébrale causée par des mutations génétiques somatiques (présentes dans quelques cellules du cerveau seulement). La région affectée, qui peut varier de localisation en fonction des individus, est désorganisée et présente des cellules cytomégaliques, c’est-à-dire de taille anormalement grande. La DCF2 entraine des épilepsies résistantes aux traitements médicamenteux, débutant dans l’enfance et nécessitant des interventions chirurgicales visant retirer la zone épileptogène. Cependant en fonction de la localisation de la zone à l’origine des crises, certains enfants ne peuvent être opérés.
Trouver un traitement efficace pour ces patients reste une problématique majeure, tant pour eux que pour leurs familles. Pour essayer de répondre à ce besoin, Stéphanie Baulac et Sara Baldassari ont mené une étude sur des tissus postopératoires de patients porteurs de mutations connues dans les gènes RHEB, mTOR ou PIK3CA.
A l’aide de plusieurs techniques, notamment le séquençage des ARN de cellule unique, les chercheuses et chercheurs ont découvert que les mutations présente dans la région affectée étaient présentes dans plusieurs types cellulaires du cerveau, principalement les neurones glutamatergiques et les astrocytes, mais aussi les interneurones GABAergiques, les oligodendrocytes et la microglie. Ceci suggère que ces anomalies génétiques apparaissent très tôt au cours du développement avant même que les cellules souches neurales se différencient.
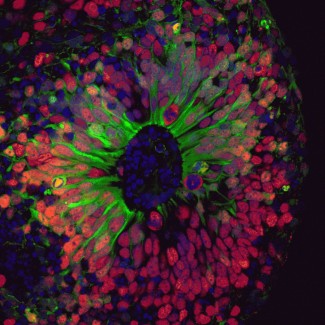
Les neurones cytomegaliques (en rose clair) présentent une forte expression des nouveaux gènes marqueurs NRGN (points orange) et STMN2 (points jaunes). Les noyaux cellulaires sont en bleu et les neurones de taille normale sont en vert.
Moins de 10% des cellules mutées sont cytomégaliques, ce qui suggère qu'elles ne représentent que la partie visible de l’iceberg. Cela laisse supposer l'existence d'autres mécanismes pathologiques non détectables avec des approches histologiques.
Identifier les mécanismes à l'origine de la maladie
Ces travaux ont permis d’identifier des mécanismes physiologiques spécifiques à l’origine de la maladie. Les neurones cytomégaliques sont hyperexcitables et présentent une altération de leurs mitochondries et de la sénescence cellulaire. Ces dysfonctionnements pourraient être à l’origine des crises d’épilepsie.
Grâce à ces résultats, de nouvelles pistes thérapeutiques émergent : cibler les mitochondries et la sénescence cellulaire pourrait ouvrir la voie à des traitements innovants pour les patients atteints de DCF2.
L'équipe "Mosaïcisme Génétique dans l'Épilepsie et les Troubles Neurodéveloppementaux" s'intéresse aux épilepsies focales et aux malformations du développement cortical.
En savoir plus