En collaboration avec une équipe Allemande de l‘Institut de Génétique Humaine de Würzburg et un consortium Européen (EuroEPINOMICS consortium), Caroline Nava, Christel Depienne et Eric LeGuern (dans le cadre d’une collaboration entre l’équipe d’Eric LeGuern et Stéphanie Baulac et celle d’Alexis Brice), en lien avec le Dr Carine Dalle, responsable de la plateforme d’électrophysiologie (IHU-A-Institut du Cerveau – ICM) viennent de découvrir qu’une mutation du gène HCN1 serait responsable d’une cause de l’épilepsie sévère survenant chez le nourrisson.
Les encéphalopathies épileptiques sont des épilepsies sévères proches du syndrome de Dravet, forme la plus connue des encéphalopathies épileptiques (EE) d’origine génétique. Elles se manifestent progressivement par une dégénérescence cognitive voire motrice.
Les mutations du gène HCN1 ont pu être identifiées à partir du séquençage des parties codantes du génome chez plusieurs enfants atteints du syndrome de Dravet. Dans la majorité des cas, ce syndrome est dû à une mutation dominante du gène SCN1A, codant pour un canal neural sodium voltage-dépendant.
Le gène HCN1, lui, permet la synthèse de sous-unités constituant un canal neural formé de 4 sous-unités, les canaux HCN, responsables d’un courant aux propriétés particulières appelé Ih. Ce courant est important pour la propagation de l’influx nerveux.
En outre, les symptômes cliniques chez les patients atteints de la mutation du gène HCN1 sont similaires à ceux observés dans le syndrome de Dravet, dans les premières années de vie. Néanmoins, il a été constaté que ces manifestations symptomatiques évoluent différemment ; les patients les plus âgés développent tous des crises d’épilepsie focale, des épisodes d’absence et des troubles du comportement importants avec des traits autistiques.
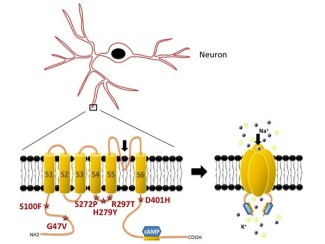
Ces recherches ont pu être menées grâce aux techniques d’électrophysiologie associées à une approche de biologie moléculaires, développées au sein de l’IHU-A-Institut du Cerveau – ICM, qui permettent d’aborder aujourd’hui, la physiopathologie d’un nombre croissant de maladies qui affectent la transmission nerveuse et conduisent à des pathologies invalidantes du système nerveux.
L’identification de mutations du gène HCN1 confirme le rôle crucial des canaux HCN et du courant ih dans les mécanismes impliqués dans les épilepsies chez l’homme. La découverte d’une nouvelle cause génétique responsable d’encéphalopathie épileptique sévère va par ailleurs permettre d’offrir un nouveau diagnostic et une explication de la maladie aux familles.

Sources
Nava C, Dalle C, Rastetter A, Striano P, de Kovel C, Nabbout R, Cancès C, Ville D, Brilstra EH, Gobbi G, Raffo E, Bouteiller D, Marie Y, Trouillard O, Robbiano A, Keren B, Agher D, Roze E, Lesage S, Nicolas A, Brice A, Baulac M, Vogt C, El Hajj N, Schneider E, Suls A, Weckhuysen S, Gormley P, Lehesjoki AE, De Jonghe P, Helbig I, Baulac S, Zara F, Koeleman B, EuroEPINOMICS consortium Haaf T, LeGuern E, and Depienne C. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nature Genetics.