Giovanni Stevanin, chercheur à l’Institut du Cerveau et de la Moelle épinière, et ses collaborateurs ont identifié une mutation récurrente dans un nouveau gène responsable d’ataxie cérébelleuse qui code un canal calcique exprimé dans certains neurones du cervelet.
De nombreux médicaments régulant l’activité des canaux calciques sont largement utilisés et cette découverte pourrait mener à des avancées thérapeutiques pour ces maladies rares. Cette étude est publiée dans la revue The American Journal of Human Genetics.
Les ataxies cérébelleuses sont des maladies dégénératives touchant le cervelet, structure majeure du système nerveux central, intervenant dans l’équilibre et la coordination. Les patients atteints présentent des symptômes très invalidants, handicapant la marche ou la réalisation des mouvements. Certaines formes d’ataxie cérébelleuse sont d’origine génétique. Même si plus de 50 gènes impliqués ont été découverts, la cause reste encore inconnue chez plus de 40% des patients.
Grâce à la combinaison de nouvelles technologies de séquençage à haut débit et de cartographie du génome à grande échelle, Giovanni Stevanin et Marie Coutelier à l’Institut du Cerveau et de la Moelle épinière, Philippe Lory à l’Institut de Génétique Fonctionnelle à Montpellier et leurs collaborateurs ont identifié une mutation récurrente (affectant 3 familles indépendantes) dans un nouveau gène, CACNA1G, responsable de formes autosomiques dominantes d’ataxie cérébelleuse. Ce gène code un canal (Cav3.1) laissant passer les ions calcium dans certains neurones du cervelet, les cellules de Purkinje. La mutation affecte une partie critique de la protéine qui est responsable de la détection des changements de potentiel de la membrane cellulaire qui entraînent l’ouverture ou la fermeture du canal. La mutation décrite rend le canal moins sensible, c’est-à-dire que son activité est globalement diminuée.
Ces résultats attirent à nouveau l’attention sur l’importance majeure des canaux ioniques dans les processus menant à l’ataxie cérébelleuse. De façon intéressante, des mutations qui augmentent l’activité de ce canal ont été décrites comme des facteurs de risque d’épilepsie ce qui constitue un lien à approfondir pour mieux comprendre ces deux familles de pathologies. L’association fréquente des syndromes ataxiques et épileptiques liés aux mutations des canaux ioniques illustre leur implication simultanée dans plusieurs processus neuronaux complexes.
De nombreux médicaments régulant l’activité des canaux pour le calcium sont largement utilisés de nos jours et cette découverte pourrait mener à des avancées thérapeutiques pour ces maladies relativement orphelines.
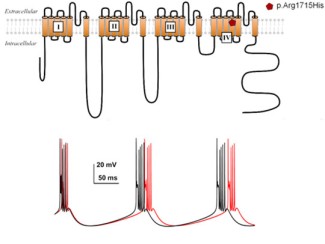
Figure : (haut) : schéma du canal enchâssé dans la membrane plasmique montrant la position de la mutation récurrente identifiée dans les familles avec ataxie. (bas): simulation des bouffées de décharge des potentiels d’action des neurones des noyaux profonds du cervelet à l’aide du modèle NEURON montrant une différence dans l’activité de décharge en présence de la mutation (rouge) comparée au canal sauvage (noir).
Sources
https://www.cell.com/ajhg/fulltext/S0002-9297(15)00371-7?_returnURL=htt…
Marie Coutelier et al., American Journal Human Genetics. 2015.