Une étude européenne conduite par Giulia Coarelli (Inserm) dans l’équipe d’Alexandra Durr (Sorbonne Université/APHP) et de Giovanni Stevanin (Inserm/EPHE) à l’Institut du Cerveau – ICM, met en évidence le lien entre certains symptômes parfois observés dans la paraplégie spastique de type 7 et certaines mutations génétiques spécifiques. Les résultats sont publiés dans la revue Neurology.
Les paraplégies spastiques héréditaires (HSP) sont un groupe de maladies très hétérogènes, d’un point de vue clinique comme génétique avec de nombreux gènes connus comme étant impliqués. Ces maladies sont héréditaires et causées par la mutation d’un gène. 79 loci ont été identifiés à l’heure actuelle et leur nombre est en constante augmentation.
Parmi eux, SPG7, responsable de la paraplégie spastique de type 7 est une de formes le plus fréquentes à transmission autosomique récessive. Comme dans les autres paraplégies spastiques, le principal symptôme de la maladie est une spasticité et une faiblesse des membres inférieurs conduisant à de sévères troubles de la marche. En revanche, un certain nombre de patients présentent également des symptômes d’ataxie cérébelleuse. Ils peuvent ainsi présenter initialement une ataxie cérébelleuse et n’être diagnostiqués d’une paraplégie spastique de type 7 qu’a posteriori. Ces patients présentent-ils des différences génétiques par rapport aux patients ne présentant que des signes de paraplégie spastique ?
Une étude collaborative européenne, réunissant des centres français, allemand, belge, néerlandais et italien, et conduite par Giulia Coarelli (Inserm) dans l’équipe d’Alexandra Durr (Sorbonne Université/APHP) et Giovanni Stevanin (Inserm/EPHE) à l’Institut du Cerveau – ICM, a permis de collecter les données de 241 patients atteints de cette pathologie, puis a cherché à étudier cette corrélation entre la génétique des patients et leurs signes cliniques.
Ils mettent en évidence qu’une mutation spécifique (p.Ala510Val), la plus fréquemment retrouvée chez les patients, est associée à un début de la maladie plus tardif et à l’apparition des symptômes d’ataxie cérébelleuse au début de la maladie. De plus, les mutations du gène SPG7 conduisant à une perte de fonction de la protéine associée entraine un tableau clinique plus complexe et sévère caractérisé par un prédominant syndrome pyramidal, une atrophie optique et une atteinte de la sensibilité profonde.
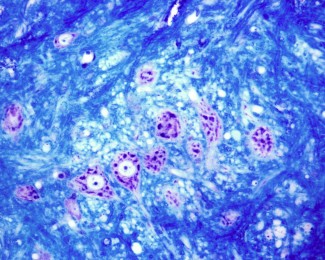
Fait rare, les chercheurs ont pu étudier le cerveau d’une patiente atteinte de SPG7. Deux études d’anatomie pathologique ont été menées dans cette pathologie jusqu’à présent. Les résultats montrent une atrophie du noyau dentelé du cervelet, du cortex cérébelleux et des cordons pyramidaux au niveau médullaire, ainsi qu’une perte neuronale de la substance noire sans atteinte du cortex moteur primaire.
La paraplégie spastique de type 7 se transmet en principe sur mode de transmission dit autosomique récessif, c’est-à-dire que les deux copies du gène doivent être mutées pour que la maladie s’exprime. L’étude a pourtant mis en évidence 12 familles pour lesquelles le mode de transmission serait autosomique dominant, c’est-à-dire qu’une seule copie du gène muté suffirait à déclencher la maladie, soulevant de nombreuses questions sur le fond génétique de SPG7.
Dans leur ensemble, ces résultats apportent des informations importantes sur la corrélation entre le génotype et le phénotype dans la paraplégie spastique de type 7 et confirme SPG7 comme une cause fréquente d’ataxie cérébelleuse. La mise en évidence d’un possible mode de transmission autosomique dominant a également des implications importantes pour le conseil génétique aux patients et à leurs familles.
Sources
https://pubmed.ncbi.nlm.nih.gov/31068484/
Coarelli G, Schule R, van de Warrenburg BPC, De Jonghe P, Ewenczyk C, Martinuzzi A, Synofzik M, Hamer EG, Baets J, Anheim M, Schöls L, Deconinck T, Masrori P, Fontaine B, Klockgether T, D’Angelo MG, Monin ML, De Bleecker J, Migeotte I, Charles P, Bassi MT, Klopstock T, Mochel F, Ollagnon-Roman E, D’Hooghe M, Kamm C, Kurzwelly D, Papin M, Davoine CS, Banneau G, Tezenas du Montcel S, Seilhean D, Brice A, Duyckaerts C, Stevanin G, Durr A. Neurology. 2019 May 8