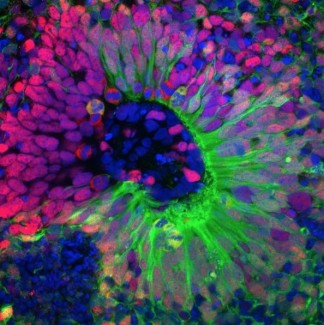
Pourquoi une même mutation génétique provoque-t-elle une malformation cérébrale sévère chez certains patients, et pas chez d’autres ? Des chercheurs de l’équipe MOSAIC, à l'Institut du Cerveau, ont généré des organoïdes corticaux humains porteurs de mutations du gène DEPDC5 afin de modéliser la dysplasie corticale focale – une malformation cérébrale responsable d'épilepsies pharmaco-résistantes chez l'enfant. Dans une nouvelle étude publiée dans Brain, ils montrent qu’une inactivation biallélique de ce gène est nécessaire pour reproduire les caractéristiques des dysplasies focales corticales, et identifient les mécanismes clés du processus pathologique.
On estime que près de 700 000 personnes souffrent d’épilepsie en France. Près de la moitié de ces patients ont moins de 20 ans et, pour un tiers d’entre eux, les médicaments disponibles demeurent inefficaces. Dans certains cas, une anomalie structurelle du cerveau est à l’origine des crises : la dysplasie corticale focale de type II (FCDII), une malformation liée au développement qui perturbe l’organisation des cellules nerveuses du cortex. Pour ces patients, l'unique recours est souvent la chirurgie – une ablation de la zone épileptogène – avec des résultats variables et des risques non négligeables.
La FCDII appartient à un groupe de maladies appelées « mTORopathies », provoquées par des mutations dans des gènes qui contrôlent la croissance et la différenciation cellulaires via la voie de signalisation mTOR. Ces mutations, qui peuvent toucher différents types cellulaires du cerveau en développement, comme les neurones, les astrocytes, ou les oligodendrocytes, créent une mosaïque : certaines cellules portent la mutation, d'autres non.
Un modèle expérimental inédit : des mini-cerveaux mosaïques
Pour percer les mécanismes de la FCDII, l'équipe MOSAIC à l'Institut du Cerveau a développé une approche expérimentale originale. Les chercheurs ont prélevé des cellules sanguines sur un jeune patient porteur d'une mutation du gène DEPDC5 – l'un des principaux gènes impliqués dans la FCDII – et sur son frère non atteint, qui a servi de contrôle. Ces cellules ont été reprogrammées en cellules souches pluripotentes (iPSC), puis différenciées en organoïdes corticaux humains, de petites structures tridimensionnelles de quelques millimètres de long qui imitent le développement du cerveau.
« Il existait déjà des modèles murins de cette maladie, mais ils ne reproduisaient pas parfaitement les caractéristiques des patients », explique Marina Maletic, première autrice de l’étude, qui a travaillé sur ce projet dans le cadre de sa thèse. « Le développement cortical humain présente des spécificités qu’on ne trouve pas chez la souris. Nous avons donc créé des organoïdes mosaïques humains, obtenus en mixant des cellules portant deux copies mutées du gène DEPDC5 avec des cellules n'en portant qu'une seule. Cela nous permet de reconstituer la situation génétique observée dans le cerveau des patients. »
L’équipe a comparé trois types d'organoïdes au cours de six mois de développement : des organoïdes contrôles (sains), des organoïdes hétérozygotes (une seule copie du gène mutée) et les fameux organoïdes mosaïques. Cette comparaison longitudinale a permis d'identifier les anomalies nécessitant l'inactivation complète du gène pour apparaître.
La nécessité d'un « double-hit »
Les résultats de l’étude confirment qu’un modèle théorique bien connu, notamment en génétique des cancers, s’applique également à la dysplasie corticale focale : le modèle du double-hit, formulé par le généticien américain Alfred G. Knudson en 1971, selon lequel deux « coups » successifs à l’ADN sont nécessaires pour provoquer un cancer.
Dans la FCDII, une première mutation héritée du gène DEPDC5 ne suffit pas à provoquer la maladie. C’est l’acquisition d'une deuxième mutation somatique – dans une cellule précise, à un moment précis du développement cérébral – qui déclenche le processus pathologique.
En effet, seuls les organoïdes mosaïques ont reproduit les caractéristiques de la maladie, telles que des neurones anormalement grands, des neurofilaments accumulés, et une hyperactivité de la voie mTOR.
« La perte totale de DEPDC5 dans une fraction de cellules nerveuses est nécessaire et suffisante pour initier la maladie. L’étendue des lésions dépend ensuite du taux de mosaïcisme, c’est-à-dire du nombre de cellules dont l’ADN a subi une seconde mutation », résume Marina Maletic.
Un développement cérébral dérégulé dès les premières semaines
Pour comprendre comment la mutation perturbe la construction du cerveau, les chercheurs ont examiné l'activité cellulaire des organoïdes à trois moments clés de leur développement – un mois, trois mois, et six mois – grâce à une technique appelée séquençage de l'ARN cellule par cellule. Celle-ci permet de déterminer quels gènes sont actifs dans chaque cellule à un instant donné et d'identifier les dérèglements invisibles au premier abord.
Le cortex cérébral humain se construit selon une chronologie très précise : certains types de neurones doivent apparaître tôt, d'autres plus tard.
« Ce calendrier était perturbé dès le premier mois, même dans les organoïdes ne portant qu'une seule copie mutée du gène : les neurones des couches supérieures du cortex apparaissaient prématurément », explique Sara Bizzotto, co-première autrice de l’étude.
À l'origine de cette accélération : la perte de DEPDC5 activait de manière anormale des gènes qui contrôlent le rythme de maturation des cellules nerveuses — en particulier ceux impliqués dans deux grandes voies de communication cellulaire, Notch et Wnt, qui régulent l'équilibre entre la multiplication des cellules souches et leur transformation en neurones matures.
Une hyperactivité neuronale mesurable
Enfin, l'enregistrement de l'activité électrique spontanée des organoïdes à six mois a mis en évidence une excitabilité pathologique du réseau neuronal dans les organoïdes mosaïques, possiblement due à des gènes liés à l’épilepsie dérégulés au sein des neurones. Ces derniers déchargeaient plus souvent et de manière plus étendue que dans les organoïdes contrôles et les hétérozygotes.
« On ne peut pas vraiment parler d’épilepsie chez un organoïde. C’est un modèle, pas un patient ! Toutefois, nous considérons cette activité électrique anormale comme un corrélat des crises épileptiques chez l’humain. Cette observation clôt notre description du processus pathologique », ajoute Marina Maletic.
De nouvelles perspectives de recherche
Outre leur contribution à la compréhension de la dysplasie corticale focale de type II, les chercheurs suggèrent que les gènes épileptiques dérégulés pourraient constituer de nouvelles cibles thérapeutiques.
Enfin, les organoïdes mosaïques ouvrent la voie à la modélisation d'autres malformations cérébrales mosaïques, dont l'étude a longtemps été entravée par l'inaccessibilité du tissu cérébral humain.
« C’est un excellent modèle qui permettra, à terme, de faire de la médecine de précision : en développant des mini-cerveaux de laboratoire à partir des cellules d’un patient, nous pourrons tester plusieurs options thérapeutiques pour déterminer laquelle lui convient le mieux », conclut-elle.
Sources
Maletic, M., Bizzotto, S. et al. Mosaic human cortical organoids model 1 mTOR-related focal cortical dysplasia 2 through DEPDC5 deletion. Brain, Mars 2026. DOI : 10.1093/brain/awag086.
Financement
Ce travail a été financé par le Conseil européen de la recherche, le programme « Investissements d'avenir », la Fondation pour la recherche médicale (FRM) et le réseau ERA-NET NEURON.
Image
Organoïde de cerveau. Crédit : Marina Maletic.
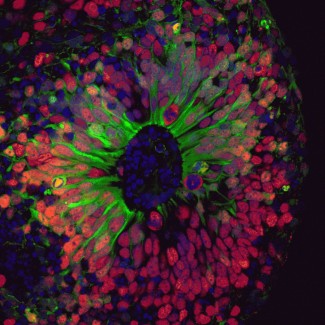
L'équipe "Mosaïcisme Génétique dans l'Épilepsie et les Troubles Neurodéveloppementaux" s'intéresse aux épilepsies focales et aux malformations du développement cortical.
En savoir plus